
ABRYSVO, poudre et solvant pour solution injectable. Vaccin du virus respiratoire syncytial (bivalent, recombinant), boîte de 1 flacon de poudre seringue préremplie ( 1 aig) de ½ mL
Dernière révision : 10/04/2025
Taux de TVA : 2.1%
Prix de vente : 196,10 €
Taux remboursement SS : 30%
Base remboursement SS : 196,10 €
Laboratoire exploitant : PFIZER
Source :

Abrysvo est indiqué pour :
La protection passive contre la maladie des voies respiratoires inférieures causée par le virus respiratoire syncytial (VRS) chez les nourrissons de la naissance jusqu'à l'âge de 6 mois à la suite de l'immunisation de la mère pendant la grossesse. Voir rubriques Posologie et mode d'administration et Propriétés pharmacodynamiques.
L'immunisation active des personnes âgées de 18 ans et plus pour la prévention de la maladie des voies respiratoires inférieures causée par le VRS.
L'utilisation de ce vaccin doit être conforme aux recommandations officielles.
Hypersensibilité aux substances actives ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Hypersensibilité et anaphylaxie
Un traitement médical approprié et une surveillance doivent toujours être disponibles en cas d'événement anaphylactique consécutif à l'administration du vaccin.
Réactions en rapport avec l'anxiété
Des réactions en rapport avec l'anxiété, notamment des réactions vaso-vagales (syncope), de l'hyperventilation ou des réactions en rapport avec le stress peuvent survenir lors de la vaccination comme réaction psychogène à l'injection avec une aiguille. Il est important que des mesures soient mises en place afin d'éviter toute blessure en cas d'évanouissement.
Maladie concomitante
La vaccination doit être reportée chez les personnes souffrant d'une maladie fébrile aiguë. Toutefois, la présence d'une infection mineure, telle qu'un rhume, ne doit pas entraîner le report de la vaccination.
Thrombopénie et troubles de la coagulation
Abrysvo doit être administré avec précaution chez les personnes présentant une thrombopénie ou tout trouble de la coagulation car un saignement ou une ecchymose peut survenir suite à une administration intramusculaire chez ces personnes.
Personnes immunodéprimées
L'efficacité et la sécurité du vaccin n'ont pas été évaluées chez les personnes immunodéprimées, notamment celles traitées par immunosuppresseurs. L'efficacité d'Abrysvo peut être plus faible chez les personnes immunodéprimées.
Personnes ayant moins de 24 semaines d'aménorrhée
Abrysvo n'a pas été étudié chez les femmes enceintes de moins de 24 semaines d'aménorrhée. Étant donné que la protection du nourrisson contre le VRS dépend du transfert des anticorps maternels à travers le placenta, Abrysvo doit être administré entre la 24e semaine et la 36e semaine d'aménorrhée (voir rubriques Posologie et mode d'administration et Propriétés pharmacodynamiques).
Limites de l'efficacité vaccinale
Comme tout vaccin, il se peut qu'une réponse immunitaire protectrice ne soit pas déclenchée après la vaccination.
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c'est-à-dire qu'il est essentiellement « sans sodium ».
Abrysvo contient du polysorbate 80. Le polysorbate 80 peut provoquer des réactions d'hypersensibilité.
Résumé du profil de sécurité
Femmes enceintes
Chez les femmes enceintes de 24 à 36 semaines d'aménorrhée, les effets indésirables les plus fréquemment rapportés étaient des douleurs au site de vaccination (41 %), des céphalées (31 %) et des myalgies (27 %). Dans la majorité des cas, les réactions locales et systémiques chez les mères participantes étaient de sévérité légère à modérée et se sont résolues dans les 2 à 3 jours suivant leur apparition.
Personnes âgées de 18 ans et plus
Chez les personnes âgées de 18 ans et plus, les effets indésirables les plus fréquemment rapportés étaient la fatigue (23 %), les céphalées (20 %), les douleurs au site de vaccination (19 %) et la myalgie (16 %). Dans la majorité des cas, les réactions ont été de sévérité légère à modérée et se sont résolues dans les 1 à 2 jours suivant leur apparition.
Tableau mentionnant les effets indésirables
La sécurité de l'administration d'une dose unique d'Abrysvo à des femmes enceintes de 24 à 36 semaines d'aménorrhée (n = 3 698) et à des personnes âgées de 18 ans et plus (n = 20 275) a été évaluée dans des essais cliniques.
Les effets indésirables sont répertoriés selon les catégories de fréquence suivantes :
Très fréquent (≥ 1/10) ;
Fréquent (≥ 1/100, < 1/10) ;
Peu fréquent (≥ 1/1 000, < 1/100) ;
Rare (≥ 1/10 000, < 1/1 000) ;
Très rare (< 1/10 000) ;
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Les effets indésirables rapportés sont répertoriés par classe de systèmes d'organes, par ordre décroissant de gravité.
Tableau 1 Effets indésirables suite à l'administration d'Abrysvo
|
Classe de systèmes d'organes |
Effets indésirables Personnes enceintes âgées de ≤ 49 ans |
Effets indésirables Personnes âgées de ≥ 18 ans |
|
Affections hématologiques et du système lymphatique |
|
|
|
Lymphadénopathie |
Rare |
Rare |
|
Affections du système immunitaire |
|
|
|
Anaphylaxie |
|
Très rare |
|
Réactions d'hypersensibilité (incluent éruption cutanée, urticaire) |
Rare |
Rare |
|
Affections du système nerveux |
|
|
|
Céphalées |
Très fréquent |
Très fréquent |
|
Syndrome de Guillain-Barré |
|
Très rare |
|
Affections musculosquelettiques et du tissu conjonctif |
|
|
|
Myalgie |
Très fréquent |
Très fréquent |
|
Arthralgie |
|
Fréquent |
|
Troubles généraux et anomalies au site d'administration |
|
|
|
Fatigue |
|
Très fréquent |
|
Douleurs au site de vaccination |
Très fréquent |
Très fréquent |
|
Rougeur au site de vaccination |
Fréquent |
Fréquent |
|
Gonflement au site de vaccination |
Fréquent |
Fréquent |
|
Pyrexie |
|
Peu fréquent |
|
Prurit au site de vaccination |
|
Rare |
|
Ecchymose au site de vaccination |
|
Rare |
|
Hématome au site de vaccination |
|
Rare |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr
INFORMER IMMEDIATEMENT LE MEDECIN en cas de :
- Picotements et faiblesse des membres, pouvant aller jusqu'à la paralysie partielle ou complète du corps.
- Gonflement du visage, des lèvres, de la langue ou de la gorge,
difficultés pour respirer ou avaler et sensations
vertigineuses.
Grossesse
Les données sur les femmes enceintes (plus de 4 000 expositions) n'indiquent aucune malformation ni aucune toxicité fœtale/néonatale.
Les résultats des études effectuées chez l'animal portant sur Abrysvo n'indiquent pas d'effets délétères directs ou indirects en ce qui concerne la toxicité pour la reproduction (voir rubrique Données de sécurité préclinique).
Dans une étude de phase III (Étude 1), les effets indésirables maternels signalés dans le mois suivant la vaccination étaient similaires dans le groupe Abrysvo (14 %) et dans le groupe placebo (13 %).
Aucun signal de sécurité n'a été détecté chez les nourrissons jusqu'à l'âge de 24 mois. L'incidence des événements indésirables signalés dans le mois suivant la naissance chez les nourrissons était similaire dans le groupe Abrysvo (38 %) et dans le groupe placebo (35 %). Les anomalies majeures à la naissance, évaluées dans le groupe Abrysvo comparé au placebo, incluaient naissance prématurée (207(6 %) et 172 (5 %), respectivement), faible poids à la naissance (186 (5 %) et 158 (4 %), respectivement) et anomalies congénitales (205 (6 %) et 245 (7 %), respectivement).
Allaitement
On ne sait pas si Abrysvo est excrété dans le lait maternel. Aucun effet indésirable d'Abrysvo n'a été mis en évidence chez les nouveau-nés allaités par des mères vaccinées.
Fertilité
Aucune donnée n'est disponible concernant l'effet d'Abrysvo sur la fertilité humaine.
Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la fertilité féminine (voir rubrique Données de sécurité préclinique).
Abrysvo peut être administré de manière concomitante avec :
- les vaccins de la grippe saisonnière, à dose standard avec adjuvant ou à dose élevée sans adjuvant ;
- les vaccins à ARNm contre la COVID-19, avec ou sans administration concomitante d'un vaccin contre la grippe à dose élevée sans adjuvant.
Un intervalle minimum de deux semaines est recommandé entre l'administration d'Abrysvo et l'administration du vaccin diphtérie-tétanos-coqueluche acellulaire (dTca). Il n'y a eu aucun problème de sécurité lorsqu'Abrysvo a été administré en même temps que le dTca à des femmes non enceintes en bonne santé. Les réponses immunitaires au VRS A, au VRS B, à la diphtérie et au tétanos lors de la co-administration étaient non inférieures à celles après une administration séparée. Cependant, les réponses immunitaires aux composants de la coqueluche ont été plus faibles lors de la co-administration par rapport à l'administration séparée et n'ont pas atteint le critère de non-infériorité. La pertinence clinique de cette observation est inconnue.
Posologie
Femmes enceintes
Une dose unique de 0,5 mL doit être administrée entre la 24e et la 36e semaine d'aménorrhée (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacodynamiques).
Personnes âgées de 18 ans et plus
Une dose unique de 0,5 mL doit être administrée.
La nécessité d'une revaccination n'a pas été établie.
Population pédiatrique
La sécurité et l'efficacité d'Abrysvo chez les enfants (de la naissance à moins de 18 ans) n'ont pas encore été établies. Des données limitées sont disponibles chez les adolescentes enceintes et leurs nourrissons (voir rubrique Propriétés pharmacodynamiques).
Mode d'administration
Abrysvo doit être injecté par voie intramusculaire dans la région deltoïdienne de la partie supérieure du bras.
Le vaccin ne doit pas être mélangé avec d'autres vaccins ou médicaments.
Pour les instructions concernant la reconstitution et la manipulation du médicament avant administration, voir la rubrique Précautions particulières d’élimination et de manipulation.
Durée de conservation :
3 ans
Le flacon non ouvert est stable pendant 5 jours lorsqu'il est conservé à des températures comprises entre 8 °C et 30 °C. À la fin de cette période, Abrysvo doit être utilisé ou éliminé. Ces informations sont destinées à guider les professionnels de santé en cas d'excursions de température temporaires uniquement.
Après reconstitution
Abrysvo doit être administré immédiatement après reconstitution ou dans les 4 heures qui suivent s'il est conservé à une température comprise entre 15 °C et 30 °C. Ne pas congeler.
La stabilité physico-chimique en cours d'utilisation a été démontrée pendant 4 heures à une température comprise entre 15 °C et 30 °C. D'un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d'utilisation non immédiate, les durées et les conditions de conservation avant utilisation relèvent de la responsabilité de l'utilisateur.
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 ºC et 8 ºC).
Ne pas congeler. Jeter la boîte si elle a été congelée.
Pour les conditions de conservation après reconstitution du médicament, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Un surdosage avec Abrysvo est peu probable en raison de la présentation en dose unique.
Il n'existe pas de traitement spécifique en cas de surdosage avec Abrysvo. En cas de surdosage, la personne doit faire l'objet d'une surveillance et recevoir un traitement symptomatique approprié.
Classe pharmacothérapeutique : vaccins, autres vaccins viraux, code ATC : J07BX05
Mécanisme d'action
Abrysvo contient deux antigènes F du VRS recombinants stabilisés en forme préfusion représentant les sous-groupes VRS-A et VRS-B. La protéine F en forme préfusion est la cible principale des anticorps neutralisants qui bloquent l'infection par le VRS. Après administration intramusculaire, les antigènes F en forme préfusion provoquent une réponse immunitaire qui protège contre la maladie des voies respiratoires inférieures (MVRI) associée au VRS.
Chez les nourrissons nés de mères vaccinées avec Abrysvo entre la 24e semaine et la 36e semaine d'aménorrhée, la protection contre la MVRI associée au VRS est due au transfert transplacentaire d'anticorps neutralisant le VRS. Les adultes âgés de 18 ans et plus sont protégés par une immunisation active.
Efficacité clinique
Nourrissons de la naissance à l'âge de 6 mois par immunisation active des femmes enceintes
L'Étude 1 était une étude de phase III, multicentrique, randomisée (1/1), en double aveugle, contrôlée contre placebo, visant à évaluer l'efficacité d'une dose unique d'Abrysvo dans la prévention de la MVRI associée au VRS chez les nourrissons nés de femmes enceintes vaccinées entre la 24e semaine et la 36e semaine d'aménorrhée. La nécessité d'une revaccination lors des grossesses ultérieures n'a pas été établie.
Une MVRI associée au VRS a été définie comme la confirmation d'un VRS par réaction en chaîne par polymérase à transcription inverse (RT-PCR) chez des individus suivis médicalement et présentant un ou plusieurs des symptômes respiratoires suivants : respiration rapide, faible saturation en oxygène (SpO2 < 95 %) et tirage sous-costal. Une MVRI sévère associée au VRS a été définie comme une MVRI associée au VRS répondant en outre à au moins l'un des critères suivants : respiration très rapide, faible saturation en oxygène (SpO2 < 93 %), supplémentation en oxygène à haut débit par canule nasale ou ventilation mécanique, admission en USI pendant > 4 heures et/ou absence de réponse/inconscience.
Au cours de cette étude, 3 711 femmes enceintes, dans le cadre de grossesses simples et sans complication, ont été randomisées dans le groupe Abrysvo et 3 709 dans le groupe placebo.
L'efficacité vaccinale (EV) a été définie comme la réduction du risque relatif du critère d'évaluation dans le groupe Abrysvo par rapport au groupe placebo pour les nourrissons nés de femmes enceintes ayant reçu le médicament expérimental attribué. Lors de l'analyse principale, deux critères d'efficacité principaux ont été évalués en parallèle : la MVRI sévère suivie médicalement due au VRS et la MVRI suivie médicalement due au VRS, survenant dans les 90, 120, 150 ou 180 jours après la naissance.
Parmi les femmes enceintes ayant reçu Abrysvo, 65 % étaient blanches, 20 % étaient noires ou afroaméricaines et 29 % étaient hispaniques/latino-américaines. L'âge médian était de 29 ans (intervalle : 16 - 45 ans) ; 0,2 % des participantes avaient moins de 18 ans et 4,3 % moins de 20 ans. L'âge gestationnel médian au moment de la vaccination était de 31 semaines d'aménorrhée et 2 jours (de 24 semaines d'aménorrhée et 0 jour à 36 semaines d'aménorrhée et 4 jours). L'âge gestationnel médian du nourrisson à la naissance était de 39 semaines d'aménorrhée et 1 jour (de 27 semaines d'aménorrhée et 3 jours à 43 semaines d'aménorrhée et 6 jours).
L'efficacité vaccinale est présentée dans les tableaux 2 et 3.
Tableau 2 Efficacité vaccinale d'Abrysvo contre la MVRI sévère suivie médicalement causée par le VRS chez les nourrissons de la naissance à l'âge de 6 mois par immunisation active des femmes enceintes — Étude 1
|
Période |
Abrysvo Nombre de cas N = 3 495 |
Placebo Nombre de cas N = 3 480 |
EV (%) (IC)a |
|
90 jours |
6 |
33 |
81,8 (40,6 ; 96,3) |
|
120 jours |
12 |
46 |
73,9 (45,6 ; 88,8) |
|
150 jours |
16 |
55 |
70,9 (44,5 ; 85,9) |
|
180 jours |
19 |
62 |
69,4 (44,3 ; 84,1) |
IC = intervalle de confiance ; EV = efficacité vaccinale
a IC à 99,5 % à 90 jours ; IC à 97,58 % aux intervalles ultérieurs
Tableau 3 Efficacité vaccinale d'Abrysvo contre la MVRI suivie médicalement causée par le VRS chez les nourrissons de la naissance à l'âge de 6 mois par immunisation active des femmes enceintes — Étude 1
|
Période |
Abrysvo Nombre de cas N = 3 495 |
Placebo Nombre de cas N = 3 480 |
EV (%) (IC)a |
|
90 jours |
24 |
56 |
57,1 (14,7 ; 79,8) |
|
120 jours |
35 |
81 |
56,8 (31,2 ; 73,5) |
|
150 jours |
47 |
99 |
52,5 (28,7 ; 68,9) |
|
180 jours |
57 |
117 |
51,3 (29,4 ; 66,8) |
IC = intervalle de confiance ; EV = efficacité vaccinale
a IC à 99,5 % à 90 jours ; IC à 97,58 % aux intervalles ultérieurs
Une analyse post-hoc de l'EV selon l'âge gestationnel maternel a été conduite. Pour les cas sévères de MVRI suivis médicalement survenus dans les 180 jours, l'EV était de 57,2 % (IC à 95 % : 10,4 ; 80,9) pour les femmes vaccinées tôt au cours de leur grossesse (24 à < 30 semaines d'aménorrhée) et de 78,1 % (IC à 95 % : 52,1 ; 91,2) pour les femmes vaccinées plus tard au cours de la fenêtre éligible de leur grossesse (30 à 36 semaines d'aménorrhée). Pour les cas de MVRI suivis médicalement survenus dans les 180 jours, l'EV était de 30,9 % (IC à 95 % : -14,4 ; 58,9) pour les femmes vaccinées tôt au cours de leur grossesse (24 à < 30 semaines d'aménorrhée) et de 62,4 % (IC à 95 % : 41,6 ; 76,4) pour les femmes vaccinées plus tard au cours de la fenêtre éligible de leur grossesse (30 à 36 semaines d'aménorrhée).
Immunisation active des personnes âgées de 60 ans et plus
L'Étude 2 était une étude de phase III, multicentrique, randomisée, en double aveugle, contrôlée contre placebo visant à évaluer l'efficacité d'Abrysvo dans la prévention de la MVRI associée au VRS chez les personnes âgées de 60 ans et plus.
La MVRI associée au VRS a été définie comme une maladie à VRS confirmée par RT-PCR, accompagnée de deux ou trois symptômes respiratoires ou plus, nouveaux ou augmentés, mentionnés ci-après, apparaissant dans les 7 jours après le premier symptôme, et durant plus d'un jour au cours du même épisode de la maladie : toux, respiration sifflante, production d'expectorations, essoufflement ou tachypnée (≥ 25 respirations/min ou augmentation de 15 % par rapport à la valeur initiale au repos).
Les participants ont été randomisés (1 :1) pour recevoir Abrysvo (n = 18 487) ou un placebo (n = 18 479). Le recrutement a été stratifié par âge, 60 - 69 ans (63 %), 70 - 79 ans (32 %) et ≥ 80 ans (5 %). Les personnes présentant des maladies chroniques sous-jacentes stables ont été éligibles pour cette étude et 52 % des participants présentaient au moins 1 maladie préspécifiée ; 16 % des participants atteints de maladies cardio-pulmonaires chroniques stables telles que l'asthme (9 %), la bronchopneumopathie chronique obstructive (7 %) ou l'insuffisance cardiaque congestive (2 %) ont été recrutés. Les personnes immunodéprimées n'étaient pas éligibles.
L'objectif principal était l'évaluation de l'efficacité vaccinale (EV), définie comme la réduction du risque relatif de premier épisode de MVRI associée au VRS dans le groupe Abrysvo par rapport au groupe placebo au cours de la première saison de VRS.
Parmi les participants ayant reçu Abrysvo, 51 % étaient des hommes et 80 % étaient blancs, 12 % étaient noirs ou afro-américains et 42 % étaient hispaniques/latino-américains. L'âge médian des participants était de 67 ans (intervalle : 59 - 95 ans).
À la fin de la première saison de VRS, l'analyse a démontré une efficacité statistiquement significative d'Abrysvo pour la réduction de la MVRI associée au VRS accompagnée de ≥ 2 symptômes et de ≥ 3 symptômes.
Les informations relatives à l'efficacité vaccinale à la fin de la première saison de VRS (durée médiane du suivi : 7,4 mois) sont présentées dans le tableau 4.
Tableau 4 Efficacité vaccinale d'Abrysvo contre la maladie à VRS — immunisation active des personnes âgées de 60 ans et plus — Étude 2
|
Critère d'efficacité |
|
Abrysvo |
Placebo |
EV (%) (IC à 95 %) |
||
|
|
|
N |
n |
N |
n |
|
|
Premier épisode de MVRI associée au VRS accompagnée de ≥ 2 symptômesa |
Total |
18 058 |
15 |
18 076 |
43 |
65,1 (35,9 ; 82,0) |
|
Participants âgés de 60 à 69 ans |
11 305 |
10 |
11 351 |
25 |
60,0 (13,8 ; 82,9) |
|
|
Participants âgés de 70 à 79 ans |
5 750 |
4 |
5 742 |
12 |
66,7 (-10,0 ; 92,2) |
|
|
Participants présentant ≥ 1 maladie sous-jacente significative |
9 377 |
8 |
9 432 |
22 |
63,6 (15,2 ; 86,0) |
|
|
Premier épisode de MVRI associée au VRS accompagnée de ≥ 3 symptômesb |
Total |
18 058 |
2 |
18 076 |
18 |
88,9 (53,6 ; 98,7) |
|
Participants âgés de 60 à 69 ans |
11 305 |
2 |
11 351 |
11 |
81,8 (16,7 ; 98,0) |
|
|
Participants âgés de 70 à 79 ans |
5 750 |
0 |
5 742 |
4 |
100 (-51,5 ; 100,0) |
|
|
Participants présentant ≥ 1 maladie sous-jacente significative |
9 377 |
2 |
9 432 |
11 |
81,8 (16,7 ; 98,0) |
|
IC = intervalle de confiance ; VRS = virus respiratoire syncytial ; EV = efficacité vaccinale
N = nombre de participants ; n = nombre de cas
a Dans une analyse exploratoire pour le sous-groupe A du VRS (Abrysvo n = 3, placebo n = 16), l'EV était de 81,3 % (IC : 34,5 ; 96,5) ; et dans le sous-groupe B du VRS (Abrysvo n = 12, placebo n = 26), l'EV était de 53,8 % (IC : 5,2 ; 78,8).
b Dans une analyse exploratoire pour le sous-groupe A du VRS (Abrysvo n = 1, placebo n = 5), l'EV était de 80,0 % (IC : -78,7 ; 99,6) ; et dans le sous-groupe B du VRS (Abrysvo n = 1, placebo n = 12), l'EV était de 91,7 % (IC : 43,7 ; 99,8).
Aucune conclusion concernant l'efficacité vaccinale dans le sous-groupe incluant des participants âgés de 80 ans et plus (995 et 981 participants dans les groupes recevant, respectivement, Abrysvo et le placebo) ne peut être tirée en raison du faible nombre total de cas cumulés (7 cas de MVRI associée au VRS avec au moins 2 symptômeset 3 cas de MVRI associée au VRS avec au moins 3 symptômes).
Efficacité contre la MVRI associée au VRS sur 2 saisons de VRS chez des individus âgés de 60 ans et plus
Sur 2 saisons de VRS avec une durée médiane de suivi de 16,4 mois, l'EV contre la MVRI associée au VRS avec au moins 2 symptômes était de 58,8 % (IC à 95 % : 43,0 ; 70,6. 54 cas dans le groupe recevant Abrysvo et 131 cas dans le groupe recevant le placebo) et de 81,5 % (IC à 95 % : 63,3 ; 91,6 ; 10 cas dans le groupe recevant Abrysvo et 54 cas dans le groupe recevant le placebo) avec au moins 3 symptômes. L'EV contre la MVRI associée au VRS causé par le VRS-A et le VRS-B était, respectivement, de 66,3 % (IC à 95 % : 47,2 ; 79,0) et de 50,0 % (IC à 95 % : 18,5 ; 70,0) pour les cas avec au moins 2 symptômes de MVRI, et, respectivement, de 80,6 % (IC à 95 % : 52,9 ; 93,4) et de 86,4 % (IC à 95 % : 54,6 ; 97,4) pour les cas avec au moins 3 symptômes de MVRI.
Sur 2 saisons de VRS, les analyses de l'EV en fonction de l'âge et des maladies sous-jacentes significatives dans les sous-groupes étaient cohérentes avec l'EV mesurée à la fin de la première saison de VRS et sont en faveur d'une EV cohérente parmi différents groupes d'âge et de risque.
Immunogénicité chez les personnes âgées de 18 à 59 ans
L'Étude 3 est une étude de phase III, multicentrique, randomisée, en double aveugle, contrôlée contre placebo, visant à évaluer la sécurité et l'immunogénicité d'Abrysvo chez des personnes âgées de 18 à 59 ans considérées comme à haut risque de développer une MVRI sévère causée par le VRS. L'Étude 3 a inclus des personnes présentant des troubles pulmonaires (y compris l'asthme), cardiovasculaires (à l'exception de l'hypertension isolée), rénaux, hépatiques, neurologiques, hématologiques ou métaboliques chroniques (y compris le diabète sucré et l'hyper/hypothyroïdie). Les participants ont été randomisés (2:1) pour recevoir une dose unique d'Abrysvo (n = 437) ou de placebo (n = 217).
Les caractéristiques démographiques dans l'Étude 3 étaient généralement similaires concernant l'âge et l'origine ethnique parmi les participants ayant reçu Abrysvo et ceux qui ont reçu le placebo. Cinquante-trois pour cent (53 %) étaient âgés de 18 à 49 ans et 47 % étaient âgés de 50 à 59 ans. Les groupes ayant reçu le vaccin et ayant reçu le placebo étaient similaires en ce qui concerne la présence d'au moins une affection préspécifiée, avec 53 % atteints d'au moins 1 affection pulmonaire chronique, 8 % atteints d'au moins 1 affection cardiovasculaire, 42 % atteints de diabète et 31 % atteints d'au moins 1 autre maladie (hépatique, rénale, neurologique, hématologique ou une autre maladie métabolique).
L'efficacité du vaccin chez les personnes âgées de 18 à 59 ans a été extrapolée par immunobridging de l'Étude 2, dans laquelle l'efficacité du vaccin avait été démontrée chez les personnes âgées de 60 ans et plus. Les critères de non-infériorité ont été atteints chez les personnes à haut risque âgées de 18 à 59 ans comparativement à un sous-groupe randomisé pour l'analyse de l'immunogénicité (groupe témoin externe) de personnes âgées de 60 ans et plus inclus dans l'Étude 2, concernant le ratio des moyennes géométriques des titres (MGT) d'anticorps neutralisants anti-VRS, si la limite inférieure de l'IC bilatéral à 95 % > 0,667 (marge de non-infériorité de 1,5) et la différence entre les taux de séroréponse, si la limite inférieure de l'IC bilatéral à 95 % > -10 % pour le VRS A et le VRS B.
Tableau 5 Comparaison des MGT d'anticorps neutralisants anti-VRS ajustées au modèle 1 mois après la vaccination par Abrysvo, chez les personnes âgées de 18 à 59 ans à haut risque (Étude 3) comparativement à celles âgées de 60 ans et plus (Étude 2)
|
|
Étude 3 - sujets âgés de 18 à 59 ans, à haut risque |
Étude 2- sujets ≥ 60 ans |
Comparaison ANCOVA |
||
|
Sous-groupes du VRS |
n |
MGT ajustée (IC à 95 %) |
n |
MGT ajustée (IC à 95 %) |
RMG ajusté (IC à 95 %) |
|
A |
435 |
41 097 (37 986, 44 463) |
408 |
26 225 (24 143, 28 486) |
1,57 (1,396 ; 1,759) |
|
B |
437 |
37 416 (34 278, 40 842) |
408 |
24 680 (22 504, 27 065) |
1,52 (1,333 ; 1,725) |
IC = intervalle de confiance ; RMG = ratio des moyennes géométriques ; MGT = moyenne géométrique des titres
Tableau 6 Comparaison des taux de séroréponse des titres d'anticorps neutralisants anti-VRS 1 mois après la vaccination par Abrysvo, chez les personnes âgées de 18 à 59 ans à haut risque (Étude 3) comparativement à celles âgées de 60 ans et plus (Étude 2)
|
|
Étude 3 - sujets âgés de 18 à 59 ans, à haut risque |
Étude 2 - sujets ≥ 60 ans |
Comparaison |
||
|
Sous-groupes du VRS |
n/N (%) |
IC à 95 % |
n/N (%) |
IC à 95 % |
Différence (IC à 95 %) |
|
A |
405/435 (93) |
90,3 ; 95,3 |
359/408 (88) |
84,4 ; 91,0 |
5,1 (1,2 ; 9,2) |
|
B |
408/437 (93) |
90,6 ; 95,5 |
347/408 (85) |
81,2 ; 88,4 |
8,3 (4,2 ; 12,6) |
IC = intervalle de confiance
Population pédiatrique
L'Agence européenne des médicaments a
différé l'obligation de soumettre les résultats d'études réalisées avec
Abrysvo chez les enfants âgés de 2 à moins de 18 ans dans la prévention
de la maladie des voies respiratoires inférieures causée par le VRS
(voir rubrique 4.2 pour les informations concernant l'usage
pédiatrique).
Sans objet.
Abrysvo n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études conventionnelles de toxicologie en administration répétée et des fonctions de reproduction et de développement n'ont pas révélé de risque particulier pour l'être humain.
Utilisation d'un flacon d'antigènes pour Abrysvo (poudre), d'une seringue préremplie de solvant et d'un adaptateur pour flacon
Abrysvo doit être reconstitué avant l'administration en ajoutant la totalité du contenu de la seringue préremplie de solvant au flacon contenant la poudre à l'aide de l'adaptateur pour flacon.
Le vaccin doit être reconstitué uniquement avec le solvant fourni.
Préparation pour l'administration
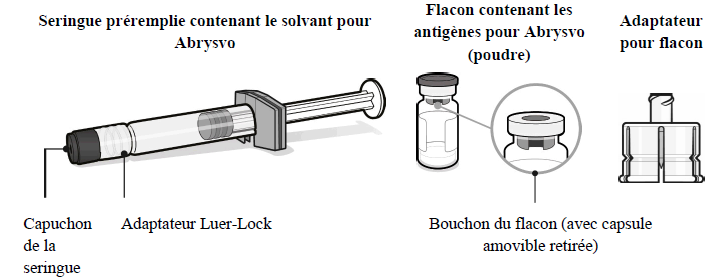
|
|
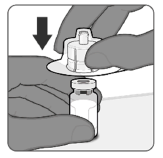
Étape 1. Fixer l'adaptateur pour flacon
|
|
|
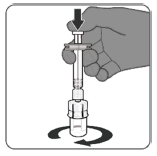
Étape 2. Reconstituer le composant en poudre (antigènes)
pour former Abrysvo
|
|
|
Étape 3. Prélever le vaccin reconstitué
|
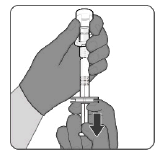
Le vaccin préparé est une solution limpide et incolore. Avant l'administration, examiner le vaccin pour vérifier l'absence de grosses particules et de coloration anormale. Ne pas utiliser si de grosses particules ou une coloration anormale sont détectées.
Utilisation du flacon d'antigènes pour Abrysvo (poudre) et du flacon de solvant
Le flacon contenant les antigènes pour Abrysvo (poudre) doit être reconstitué uniquement avec le flacon de solvant fourni pour former Abrysvo.
Préparation pour l'administration
1.
À
l'aide d'une aiguille et d'une seringue stériles, prélever la totalité du
contenu du flacon contenant le solvant et injecter tout le contenu de la
seringue dans le flacon contenant la poudre.
2. Agiter doucement le flacon en effectuant des mouvements circulaires jusqu'à dissolution complète de la poudre. Ne pas secouer.
3. Prélever 0,5 mL du flacon contenant le vaccin reconstitué.
2. Agiter doucement le flacon en effectuant des mouvements circulaires jusqu'à dissolution complète de la poudre. Ne pas secouer.
3. Prélever 0,5 mL du flacon contenant le vaccin reconstitué.
Le vaccin préparé est une solution limpide et incolore. Avant l'administration, examiner le vaccin pour vérifier l'absence de grosses particules et de coloration anormale. Ne pas utiliser si de grosses particules ou une coloration anormale sont détectées.
Élimination
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Remboursement en fonction de l'indication (JO du 15/08/2024) :
La seule indication thérapeutique ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie est la protection passive contre la maladie des voies respiratoires inférieures causée par le virus respiratoire syncytial (VRS) chez les nourrissons de la naissance jusqu'à l'âge de 6 mois à la suite de l'immunisation de la mère pendant la grossesse uniquement entre 32 et 36 semaines d'aménorrhée, selon les recommandations en vigueur de la HAS datant du 6 juin 2024.
La seule indication thérapeutique ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie est la protection passive contre la maladie des voies respiratoires inférieures causée par le virus respiratoire syncytial (VRS) chez les nourrissons de la naissance jusqu'à l'âge de 6 mois à la suite de l'immunisation de la mère pendant la grossesse uniquement entre 32 et 36 semaines d'aménorrhée, selon les recommandations en vigueur de la HAS datant du 6 juin 2024.
Poudre et solvant pour solution injectable.
La poudre est blanche.
Le solvant est un liquide limpide et incolore.
Flacon d'antigènes pour Abrysvo (poudre) et seringue préremplie de solvant
Poudre pour 1 dose dans un flacon (verre de type 1 ou équivalent) muni d’un bouchon (caoutchouc bromobutyle synthétique ou caoutchouc chlorobutyle synthétique) et d’une capsule amovible.
Solvant pour 1 dose dans une seringue préremplie (verre de type 1) munie d'un bouchon (caoutchouc chlorobutyle synthétique) et d'un capuchon d'embout (caoutchouc isoprène-bromobutyle synthétique).
Adaptateur pour flacon
Présentation
Boîte contenant 1 flacon de poudre (antigènes), 1 seringue préremplie de solvant, 1 adaptateur pour flacon avec 1 aiguille (boîte d'une dose).
Après reconstitution, une dose (0,5 mL) contient :
Antigène F du VRS du sous-groupe A stabilisé en forme préfusion1,2 60 microgrammes
Antigène F du VRS du sous-groupe B stabilisé en forme préfusion1,2 60 microgrammes
(Antigènes du VRS)
1glycoprotéine F stabilisée en forme préfusion
2produits dans des cellules ovariennes de hamster chinois par la technologie de l'ADN recombinant.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Poudre
Trométamol
Chlorhydrate de trométamol
Saccharose
Mannitol (E421)
Polysorbate 80 (E433)
Chlorure de sodium
Acide chlorhydrique (pour ajustement du pH)
Solvant
Eau pour préparations injectables